#LCSM Chat Topic 1/15 at 8PM ET: “Should the FDA Regulate Which Cancer Tests You Can Have?”
The US Food and Drug Administration (FDA) announced its intention to regulate laboratory developed tests. Under the FDA’s proposed Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs) — which treats LDTs as medical devices and healthcare providers as manufacturers — laboratories would have to submit applications for expensive premarket review for thousands of LDTs if they wish to continue offering them to patients. This could limit access to life-saving genomic testing for patients who have cancer and other conditions treatable with targeted drugs.
This Thursday, January 15, 2015, at 8 PM Eastern, the subject for #LCSM Chat will be “Should the FDA regulate which cancer tests you can have?” We invite patients, caregivers, doctors, researchers, professional societies, advocates, and regulators in all cancer communities to participate in this discussion. Your moderator will be Janet Freeman-Daily.
Our discussion topics:
- T1: What info about an LDT would give you confidence that it accurately identifies cancer or treatable mutations?
- T2: Does FDA approval ensure accuracy and usefulness of LDTs? What other info/oversight could do this?
- T3: Would FDA regulation of LDTs interfere with the practice of medicine?
- T4: Should only FDA-approved LDTs be used to guide treatment of cancer patients? Why or why not?
Background information about subject is below.
We look forward to seeing you Thursday 1/15 at 8 PM. Please be sure to include #LCSM in your tweets to participate in the chat. For more about how to participate, see our #LCSM Chat Primer.
BACKGROUND
Laboratory developed tests (LDTs) are developed, validated, performed and interpreted by trained professionals in hospital, academic, and commercial laboratories. Examples of important LDTs for lung cancer patients include blood tests (blood count, liver function, cancer biomarkers), identification of biopsied cell types (e.g., adenocarcinoma, small cell lung cancer), molecular tests (EGFR, ALK, ROS1), and genomic panels (which can test for over 200 cancer-causing gene mutations and rearrangements from one set of tissue samples). While some tests are automated, the results of these tests often depend on the judgment and skills of medical professionals such as MD pathologists or PhD scientists. Cancer-related LDTs are often developed at the request of (and in consultation with) oncologists to allow physicians to tailor treatments for their patients.
LDTs that are performed in your hospital’s lab or commercial labs (like Foundation Medicine) typically are not regulated by the FDA. However, labs are regulated and certified by the Centers for Medicare and Medicaid Services through Clinical Laboratory Improvement Amendments (CLIA), state health agencies, and organizations such as the College of American Pathologists. They also participate in programs such as proficiency testing to ensure accuracy.
Unlike LDTs, tests that are boxed and shipped to other labs and professionals contain all of the components and information necessary to perform the test outside of the laboratory in which it was designed and manufactured. Because they are manufactured by a company and not developed and validated by health professionals as part of a medical service, test kits are regulated by the FDA. The BRAF test manufactured by Roche is an example of an FDA-regulated kit.
Under the proposed framework for regulation of LDTs, the FDA would regulate LDTs just as they would medical devices such as stents, blood glucose monitors or hip replacements. Regulations would vary depending on risk categories, with tests that determine patient treatments considered as “high risk.” If this proposal were finalized, in many cases laboratories would have to pull their LDTs from their list of patient services or submit them for review by the FDA.
At first glance, FDA regulation of LDTs might seem like a good idea. The number of commercially available LDTs to detect mutations in cancer tissue has exploded from a handful in 2011 to dozens today. Some people argue we need regulations to protect vulnerable patients, citing as one example the Ovasure LDT for early detection of ovarian cancer, which the FDA forced off the market in 2008. The test aimed to detect specific proteins in the blood that, when analyzed via a mathematical algorithm, could determine whether the patient had ovarian cancer. However, the LDT was marketed before its accuracy was validated in a large group of patients. As a result, Ovasure false positives caused some women to have their ovaries removed when they did not actually have ovarian cancer. We need to prevent such things from happening, right?
Yes, we want LDTs to be as accurate and clinically useful as possible. But FDA regulation will not change the fact that ALL tests, whether an LDT or test kit, occasionally have false readings. Early in my cancer journey, a blood test said my blood glucose was 30-something (normal range is 70-120). The doctor called me late at night, concerned that I was seriously ill (if not dead). I was fine. The test result was incorrect.
The FDA held a workshop on the proposed regulations on January 8-9, 2015 (see agenda day 1 and day 2 videos). During the two days of presentations, several issues were raised :
- PACE of scientific discovery: Our knowledge of cancer-causing genes, how they affect the body, and ways of detecting them is evolving rapidly. FDA regulations move slowly; approvals usually takes years.
- VARIETY of labs producing LDTs: Some large for-profit labs that offer genomic tests might be able to afford the cost of additional personnel and fees to comply with proposed FDA regulations. Smaller labs such as those associated with hospitals might not be able to absorb the additional costs and might be forced to close.
- SCOPE of tests: Determining which LDTs to perform, validating results, and applying the results to treatment is the practice of medicine, which the FDA is prohibited from regulating. Also, the FDA seeks to regulate LDTs as medical devices, but laboratory professionals claim LDTs are not medical devices because they involve medical judgment.
Our understanding of existing oncogenes (ALK, EGFR, BRAF, etc.) and their associated targeted therapies continues to evolve even after the FDA approves companion tests to detect targetable mutations. It’s not unusual for an LDT to be developed that detects a new variation of an oncogene not detected by the FDA-approved test. Must cancer patients wait years until the FDA approves the new LDT before they can receive an effective targeted therapy? Most stage IV cancer patients can’t afford to wait that long.
Here’s an example of how pace, variety, and scope can make a difference for patients. In a presentation to the FDA on January 8, University of Colorado pathologist Dara Aisner, MD, PhD, shared the following:
“This Kaplan-Meier Curve demonstrates survival benefit for patients with metastatic melanoma treated with vemurafinib [vs dacarbazine] when they have an ‘atypical’ mutation – V600K. Of note, 34% of the V600K mutation positive patients in this cohort were classified as NEGATIVE by an FDA approved assay and were only detected using a non-FDA approved assay. … This is an example of the clinical validity that evolves rapidly with time. Determining clinical validity is the physician’s job.”
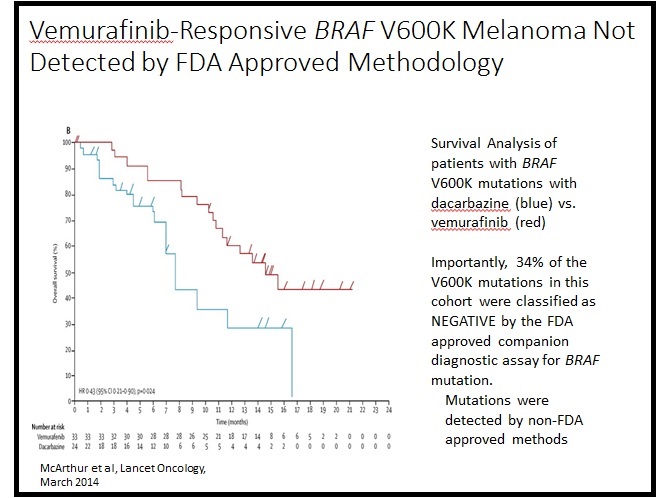
As you can see from this example, restricting the targeted therapy vemurafinib only to patients identified by the FDA-approved test would have prevented many patients from receiving effective treatment. The current FDA approval process takes years, is resource intensive, and could potentially interfere with the practice of medicine. Dr. Aisner has stated that if the FDA’s proposed regulations are enacted, her lab at the University of Colorado might have to close or at least stop providing many of its tests.
Another example: the current FDA-approved test for detecting ALK rearrangements in lung cancer is only approved for testing biopsied tumor tissue. If a patient doesn’t have sufficient biopsied tissue for testing, sometimes other sources of cells (such as fluid collected from a pleural effusion or a lymph node) can provide enough cells for ALK testing. Many labs have independently validated the test on such specimens. However, under the proposed FDA regulations, testing these alternative specimens would no longer be allowed unless a lab submits the test to the FDA and obtains its approval. As a result, some lung cancer patients would have more limited options for testing, and might require additional, potentially dangerous biopsies in order to obtain tumor tissue.
Note that the proposed regulations include an exemption for LDTs for unmet needs that would allow the use of non-FDA reviewed LDTs when no approved LDT is available for the condition. For instance, ROS1 NSCLC (my diagnosis) does not have an approved LDT, so patients could be tested with an unapproved LDT.
This proposed regulation has the potential to prevent targeted therapy treatment for thousands of patients with cancers and other diseases. We hope you’ll join us for #LCSM Chat on Thursday January 15 at 8 PM.
Comment period for the proposed FDA Framework for Regulation of Laboratory Developed Tests (LDTs) closes on February 2, 2015. Please let the FDA know what you think by submitting your comments ASAP to http://www.regulations.gov (be sure to include the docket number FDA-2011-D-0360). You can also submit comments electronically here .
REFERENCES
Overview Articles:
Opinions Divided on Proposed FDA LDT Regulations (Genetic Engineering and Biotechnology News)
To regulate or not: FDA hears arguments on medical tests (New England Center for Investigative Reporting)
Supporting the FDA’s Proposed Framework:
Advamed (medical device manufacturer’s trade association)
American Association of Cancer Research
American Society of Clinical Oncology
Journal of American Medical Association (yes)
Opposing the FDA’s Proposed Framework:
American Clinical Laboratory Association
Association for Molecular Pathology (white paper)
Joint Letter to FDA (signed by 51 organizations, societies, and laboratory directors)

Reblogged this on optimism and me and commented:
Have you ever wondered who controls the tests your can take when you’ve been diagnosed with cancer? Doesn’t seem like a big deal if it’s something your doctors say you need right? I’m sure most of us have never thought about it. If you need a certain test you should be able to take it right? Maybe. Maybe not. Read below and learn more about FDA regulations regarding cancer patients (not just lung cancer patients). Join us Thursday, 1/5 at 7 pm central for an informational, educational chat. I promise you will learn something useful!
******************